Achieving Compliant Batch Release – Sterile Parenteral Quality Control
Abstract
Demonstrating compliant final product release for a sterile parenteral batch requires the use of Quality Control (QC) instrumentation that share certain common key elements. Of course each QC instrument must complete the requirements laid down in the pharmacopoeias or GMP, but in addition each instrument should be optimized to underpin compliance, help reduce human error and maintain data integrity for the test results. This paper describes those common QC instrumentation elements and gives examples of best practice for instruments used for compliant QC batch release.
Introduction
As part of the final batch release record for sterile parenteral solutions there should be records to prove:
- The manufacturing environment (cleanroom) was in control and compliant
- The parenteral itself is compliant to the rules regarding contaminating particles
- The water used to manufacture the parenteral was in control and compliant
Cleanroom Compliance
Guidance on cleanroom compliance for sterile parenteral manufacturing can be found in the various guidelines to good manufacturing practice. The World Health Organisation2, European GMP3 and Pharmaceutical Inspection Co-operation Scheme (PIC/S)4 all agree that the air in a cleanroom must be controlled and monitored for particles ≥0.5microns and ≥5microns. The FDA’s cGMP5 document is different in that it only requires monitoring of particles ≥0.5microns. In any case, producers of sterile parenteral product must be cognisant of where each batch being produced is destined to be sold to ensure that they are being compliant to the relevant regulation(s).
The cleanroom monitoring process has traditionally been a manual process where airborne particle counters are moved around the cleanroom in a daily routine of sampling, relying on the counter operator to ensure that the test carried out at each location is correct to demonstrate the cleanroom was compliant at the time of testing. Additionally, the integrity of the final record was dependant on the operator collating all paper records from the day’s routine environmental monitoring and accurately transcribing these records into either an Excel spreadsheet, or into a secure data repository. Usually, manual calculations are also required as it is common practice to sample quite small samples at each location and then multiply the results by a factor to report the number of particles per cubic meter (m3), as is required by all of the rulebooks.
Figure 1. Routine cleanroom environmental monitoring practices are complex and fraught with opportunities for error
As can be imagined, all of this manual process can be fraught with opportunities for errors. In a modern air particle counter optimized for pharmaceutical cleanroom use, it is an expectation that the cleanroom facility routine environmental monitoring regime Standard Operating Procedure can be pre-configured inside the counter, removing the need for the operator to manually configure the sample location name, counter run time and alarms for each and every location. In addition, counters optimized for this process also automatically calculate the results per m3 and then export the results via secure file transfer, such as File Transfer Protocol (FTP), in electronic format directly to a remote file repository without any manual data manipulation required by the user/operator. Such a counter provides secure, 21CFR part 11 records to demonstrate that the cleanroom was in compliance during the manufacturing process.
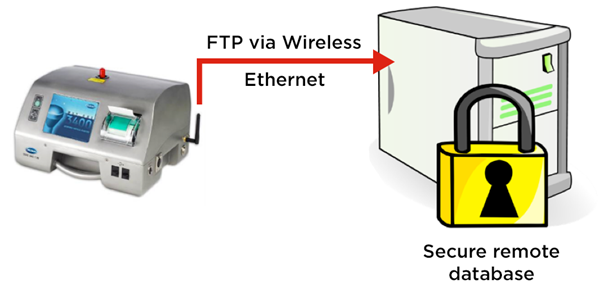
Figure 2. MET ONE 3400 particle counter exports cleanroom routine environmental monitoring electronic records securely via FTP
Final Product Particulate Contamination Compliance
The United States Pharmacopoeia chapter on therapeutic proteins, USP<787>6, suggests that sources of particles found in parenteral products can be grouped into three sources: intrinsic, extrinsic and inherent. Intrinsic particulate contamination is usually contamination from the vial or filling process due to inefficient cleaning, whereas extrinsic particulate contamination is usually introduced to the vial from the environment where the filling takes place. Inherent particles are particularly prevalent in biopharmaceutical products, where the therapeutic proteins clump together, either through adverse environmental conditions, such as bright light, temperatures or simply naturally over extended time periods.
Although largely harmonized, the rules for parenteral particulate testing do vary from country to country and from product to product. The volume of the sample to be analysed and the format that the results are reported varies from product to product, e.g. the sampling requirements for small volume parenteral product, such as vaccines, is different for that of a large volume parenteral such as an intravenous drip bag. Results must be calculated and expressed in the correct format, e.g. counts per container, or counts per mL.
Whilst general-purpose liquid particle counting instrumentation can be used for the testing of particles in parenteral products, counters that have been optimized for the application are preferable due to the wide range of complexity in the testing. Particle counters that have been optimized for this testing will have the various compendial tests built-in and will calculate a pass/fail result automatically. As QC teams tend to use their product name to describe the sample under test, optimized particle counters will allow the user to select the required test for each sample by selecting the product by name from a drop-down menu.

Figure 3. The HIAC 9703+ allows users to select final product quality testing by brand name/product name
Counters that allow the operator interface to reside on a local p.c., but store the results database automatically on a secure remote server are preferred to ensure secure, 21CFR part 11 records to demonstrate that the batch was in compliance.
Water Purity Compliance
Water is the largest raw material used in a parenteral manufacturing facility. Water quality parameters are clearly defined in all the major pharmacopoeias and are generally harmonized globally.
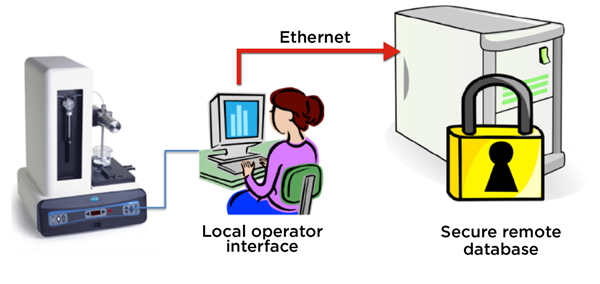
Figure 4. The HIAC 9703+ stores final product quality test result records on a remote, secure server
One major quality parameter is Total Organic Carbon (TOC). Most modern pharmaceutical-grade water systems have extremely low TOC content, frequently in the low ppb region, compared to the amount of Total Inorganic Carbon (TIC) present, typically in the low ppm region, usually caused by the increased concentration of dissolved CO2 caused through the commonly used reverse-osmosis water production process. General purpose TOC analysers that measure TIC and Total Carbon (TC) and derive the TOC level from these two measurements often struggle to accurately calculate TOC in the presence of the interfering TIC.
TC ppm (measured) – TIC ppm (measured) = TOC ppb (calculated)
Small errors in the sensor measurements for TC and TIC can lead to large variances in the calculated TOC result, sometimes producing negative TOC results where the measured and reported TIC value is slightly higher than the reported TC value.
Guidelines on instrumentation validation can be found in the International Conference on Harmonization guideline, ICH Q27. This document explicitly guides the reader to conduct an investigation on the specificity of an analytical technique to ensure that it can discriminate between compounds of closely related structures. Such guidelines can help a user validate if an analysis method designed to measure TC and TIC and calculate TOC is suitable for the water quality on their site.
As with the other instruments discussed in this paper, the ability to export results via a secure electronic transfer, such as File Transfer Protocol (FTP), to a remote 21CFR part 11 secure data repository finalizes the optimization requirements for a TOC analyser used to provide batch release data in the pharmaceutical QC application.
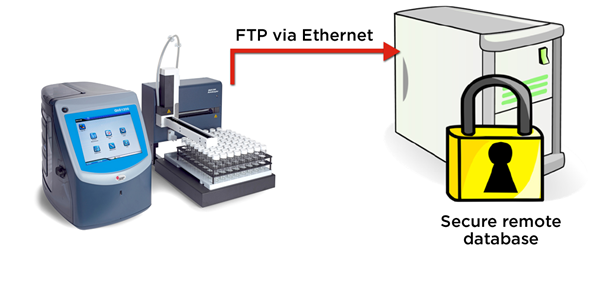
Figure 5. The QbD1200 particle counter exports WFI test records in electronic format securely via FTP
Conclusion
Pharmaceutical QC testing is complex and at the same time absolutely critical to a successful, compliant batch release. When selecting instrumentation, the QC team leader is well advised to look for instrumentation that has been optimized for pharmaceutical QC use, taking into account automated, pre-configured SOPs, built-in compendial tests and secure electronic transfer, such as File Transfer Protocol (FTP), for 21CFR part 11 electronic record retention.
References
- U.S. Department of Health and Human Services Food and Drug Administration Guidance for Industry, Part 11, Electronic Records; Electronic Signatures — Scope and Application August 2003 U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center
for Biologics Evaluation and Research (CBER) Office of Regulatory affairs (ORA) Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 USA - World Health Organisation (WHO) Good Manufacturing Practices For Sterile Pharmaceutical Products, 2009 World Health Organization, CH-1211 Geneva 27, Switzerland.
- European Commission. EudraLex. The Rules Governing Medicinal Products in the European Union. Volume 4. EU Guidelines to Good Manufacturing Practice. Medicinal products for human and veterinary use, Annex 1: Manufacture of Sterile Medicinal Products, 14th February 2008. European Commission Enterprise and Industry Directorate-General, B-1049 Bruxelles / Europese Commissie, B-1049 Brussel – Belgium.
- Pharmaceutical Inspection Co-operation Scheme, PIC/S Guide To Good Practices for The Preparation Of Medicinal Products In Healthcare Establishments, 1st April 2008, PIC/S Secretariat 14, rue du Roveray CH - 1207 Geneva Switzerland.
- Food and Drug Administration. Guidance for industry. Sterile drug products produced by aseptic processing – current good manufacturing practice, 2004. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory affairs (ORA) Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 USA
- Food and Drug Administration. USP<787> SUBVISIBLE PARTICULATE MATTER IN THERAPEUTIC PROTEIN INJECTIONS, August 1st 2014. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory affairs (ORA) Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 USA
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Validation Of Analytical Procedures: Text And Methodology Q2(R1), November 2005 [8th August 2014], http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf [8th August 2014]
- International Society for Pharmaceutical Engineering, The ISPE Good Practice Guide: Ozone Sanitization of Pharmaceutical Water Systems, First edition July 2012 http://www.ispe.org/ispe-good-practice-guides/ozone-sanitization-pharmaceutical-water-systems [14th August 2014]
- Pharmaceutical and Healthcare Sciences Society, Best Practice for Particle Monitoring in Pharmaceutical Facilities, PHSS Technical Monograph No.16, First Edition 2008, ISBN 978-1-905271-15-3
Helpful Links
-
온라인 문서
-
어플리케이션 노트
- 17-Marker, 18-Color Human Blood Phenotyping Made Easy with Flow Cytometry
- 21 CFR Part 11 Data Integrity for On-line WFI Instruments
- 8011+ Reporting Standards Feature and Synopsis
- Air Particle Monitoring ISO 21501-4 Impact
- Automated Cord Blood Cell Viability and Concentration Measurements Using the Vi‑CELL XR
- Biomek Automated NGS Solutions Accelerate Genomic Research
- Biomek i-Series Automation of the Beckman Coulter GenFind V3 Blood and Serum DNA Isolation Kit
- Preparation and purification of carbon nanotubes using an ultracentrifuge and automatic dispensing apparatus, and analysis using an analytical centrifuge system
- Viability Assessment of Cell Cultures Using the CytoFLEX
- Classifying a Small Cleanroom using the MET ONE HHPC 6+
- Clean Cabinet Air Particle Evaluation
- Recommended cleaning procedure for the exterior surface of the MET ONE 3400+
- Counting Efficiency: MET ONE Air Particle Counters and Compliance to ISO-21501
- Critical Particle Size Distribution for Cement using Laser Diffraction
- Detecting and counting bacteria with the CytoFLEX research flow cytometer: II-Characterization of a variety of gram-positive bacteria
- Efficient kit-free nucleic acid isolation uses a combination of precipitation and centrifugation separation methods
- Echo System-Enhanced SMART-Seq v2 for RNA Sequencing
- Grading of nanocellulose using a centrifuge
- Grading of pigment ink and measurement of particle diameter using ultracentrifugation / dynamic light scattering
- How to Use Violet Laser Side Scatter Detect Nanoparticle
- HRLD Recommended Volume Setting
- Particle Size Analysis Simple, Effective and Precise
- Flow Cytometric Analysis of auto-fluorescent cells found in the marine demosponge Clathria prolifera
- MET ONE Sensor Verification
- Metal colloid purification and concentration using ultracentrifugation
- Separation and purification of metal nanorods using density gradient centrifugation
- Miniaturized and High-Throughput Metabolic Stability Assay Enabled by the Echo Liquid Handler
- Minimal Sample to Sample Carry Over with the HIAC 8011+
- Modern Trends in Non‐Viable Particle Monitoring during Aseptic Processing
- Particle diameter measurement of a nanoparticle composite - Using density gradient ultracentrifugation and dynamic light scattering
- Identification of Circulating Myeloid Cell Populations in NLRP3 Null Mice
- Optimizing the HIAC 8011+ Particle Counter for Analyzing Viscous Fluids
- Particle testing in cleanroom high-pressure gas lines to ISO 14644 made easy with the MET ONE 3400 gas calibrations
- Pharma Manufacturing Environmental Monitoring
- Pharma Manufacturing Paperless Monitoring
- Analysis of plant genome sizes using flow cytometry: a case study demonstrating dynamic range and measurement linearity
- Calibrating the QbD1200 TOC Analyzer
- Detection Limit
- Vibrio Natriegens Process Scale-up
- A fully automated plate-based optimization of fed-batch culture conditions for monoclonal antibody-producing CHO cell line
- A Deeper Look at Lipid Nanoparticles
- A High-Throughput, Automated Screening Platform for IgG Quantification During Drug Discovery and Development
- Automated Research Flow Cytometry Workflow Using DURA Innovations Dry Reagent Technology with the *Biomek i7 Automated Workstation and *CytoFLEX LX Flow Cytometer
- Automating antibody titration using a CytoFLEX LX analyzer Integrated with a Biomek i7 Multichannel workstation and Cytobank streamlined data analysis
- Automated IDT Alt-R CRISPR/Cas9 Ribonucleoprotein Lipofection Using the Biomek i7 Hybrid Automated Workstation
- Monitoring Yeast Cultures with the BioLector and Multisizer 4e instruments
- Cultivation of suspended plant cells in the BioLector®
- Determination of cell death in the BioLector Microbioreactor
- Echo System-Enhanced SMART-Seq v4 for RNA Sequencing
- A Simple Guide to Selecting the Right Handheld Particle Counter for Monitoring Controlled Environments
- Linearity of the Vi-CELL BLU Cell Counter and Analyzer
- Miniaturized 16S rRNA Amplicon Sequencing with the Echo 525 Liquid Handler for Metagenomic and Microbiome Studies
- Monitoring-Synechocystis-behaviour-on-different growth-media-with-the-BioLector XT-and-Multisizer-4e
- mRNA Manufacturing with Fed Batch In Vitro Transcription
- Nanoliter Scale High-Throughput Protein Crystallography Screening with the Echo Liquid Handler
- Optimizing EV Analysis with a CytoFLEX nano flow cytometer and FCMPASS
- Preparation of Mouse Plasma Microsamples for LC-MS/MS Analysis Using the Echo Liquid Handler
- Robust and High-Throughput SARS-CoV-2 Viral RNA Detection, Research, and Sequencing Using RNAdvance Viral and the OT-2 Platform
- The Valita Aggregation Pure assay: A rapid and accurate alternative for aggregation quantification of purified monoclonal antibodies
- Accurate enumeration of phytoplankton using FCM
- Accurately measures fine bubble size and particle count
- Achieving Compliant Batch Release – Sterile Parenteral Quality Control
- Adaptive Laboratory Evolution of Pseudomonas putida in the RoboLector
- Adjustment of the pH control settings in the BioLector Pro microbioreactor
- Aerobic cultivation of high-oxygen-demanding microorganisms in the BioLector XT microbioreactor
- Comparative Analysis of DURAClone IM T-Cell Subsets Antibody Panel: Conventional vs. Spectral Flow Cytometry on CytoFLEX mosaic Spectral Detection Module
- An Analytical Revolution: Introducing the Next Generation Optima AUC
- CytoFLEX mosaic Spectral Detection Module Enables Enhanced Spectral Unmixing of White Blood Cell Populations by Extracting Multiple Autofluorescence Signatures
- Investigating the murine hepatic immune composition in diet-induced obesity using OMIP-104: Transferring an existing OMIP panel onto the CytoFLEX mosaic Spectral Detection Module
- Analyzing Mussel/Mollusk Propagation using the Multisizer 4e Coulter Counter
- Assay Assembly for Miniaturized Quantitative PCR in a 384-well Format Using the Echo Liquid Handler
- Automated 3D Cell Culture and Screening by Imaging and Flow Cytometry
- Automating a Linear Density Gradient for Purification of a Protein:Ligand Complex
- Automating Biopharma Quality Control to Reduce Costs and Improve Data Integrity
- Automating Bradford Assays
- Automating Cell-Based Processes
- Automating Cell Line Development
- Anaerobic cultivation processes of probiotic bacteria in the BioLector XT microbioreactor
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Automation of CyQuant LDH Cytotoxicity Assay using Biomek i7 Hybrid Automated Workstation to Monitor Cell Health
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Automation of protein A ELISA Assays using Biomek i7 hybrid workstation
- Avanti J-15 Centrifuge Improves Sample Protection Maximizes Sample Recovery
- The New Avanti J-15 Centrifuge Time Saving Deceleration Profile Improves Workflow Efficiency
- Avanti JXN Protein Purification Workflow
- Avoid the Pitfalls When Automating Cell Viability Counting for Biopharmaceutical Quality Control
- Basics of Multicolor Flow Cytometry
- Beer, Evaluation of Final Product and Filtration Efficiency
- Monitoring E. coli Cultures with the BioLector and Multisizer 4e Instruments
- Biomek Automated Genomic Sample Prep Accelerates Research
- Biomek i7 Hybrid Automated KAPA mRNA HyperPrep Workflow
- Biomek i-Series Automated AmpliSeq for Illumina® Library Prep Kit
- Biomek i-Series Automated Beckman Coulter Agencourt RNAdvance Blood Kit
- Biomek i-Series Automated Beckman Coulter Agencourt RNAdvance Cell
- Biomek i-Series Automated Beckman Coulter Agencourt SPRIselect for DNA Size Selection
- Biomek i-Series Automated IDT® xGen Hybridization Capture of DNA libraries on Biomek i7 Hybrid Genomics Workstation
- Biomek i-Series Automated Illumina Nextera DNA Flex Library Prep Kit
- Biomek i-Series Automated Illumina® Nextera XT DNA Library Prep Kit
- Biomek i-Series Automated Illumina TruSeq DNA PCR-Free Library Prep Kit
- Biomek i-Series Automated Illumina TruSeq® Nano DNA Library Prep Kit
- Biomek i-Series Automated Illumina TruSeq® Stranded mRNA Sample Preparation Kit Protocol
- Biomek i-Series Automated Illumina TruSeq® Stranded Total RNA Sample Preparation Kit Protocol
- Biomek i–Series Automated Illumina® TruSight Tumor 170 32 Sample Method
- Biomek i-Series Automated KAPA HyperPrep and HyperPlus Workflows
- Biomek i-Series Automated New England Biolabs NEBNext® Ultra II DNA Library Prep Kit
- Biomek i-Series Automated SurePlex PCR and VeriSeq PGS Library Prep for Illumina
- Biomek i-Series Automation of the DNAdvance Genomic DNA Isolation Kit
- Cell Counting Performance of Vi–Cell BLU Cell Viability Analyzer
- Cell Line Development – Data Handling
- Cell Line Development – Limiting Dilution
- Cell Line Development – Selection and Enrichment
- Cellular Analysis using the Coulter Principle
- Changes to GMP Force Cleanroom Re-Classifications
- Characterizing Insulin as a Biopharmaceutical Using Analytical Ultracentrifugation
- Fda guidance 21 cfr compliance guide for met one 3400 plus
- Cluster Count Analysis and Sample Preparation Considerations for the Vi-CELL BLU Cell Viability Analyzer
- Comparing Data Quality & Optical Resolution of the Next Generation Optima AUC to the Proven ProteomeLab on a Model Protein System
- Considerations of Cell Counting Analysis when using Different Types of Cells
- Consistent Cell Maintenance and Plating through Automation
- Control of Spheroid Size and Support for Productization
- Control Standards and Method Recommendations for the LS 13 320 XR
- Data-integrity-and-met-one-3400-plus-function-for-pharma
- Cydem VT Automated Clone Screening System – Generating an Antibody Standard Curve
- Cydem VT System: A Comparison to Traditional Clone Screening Platforms
- Cydem VT System Analytical Capabilities and Repeatability
- Determination of kLa values on the Cydem VT Automated Clone Screening System
- Optimize Clone Screening: Time Savings with the Cydem VT System in Monoclonal Antibody-Producing Cell Line Development Workflows
- Protein Titer Capabilities - A Comparison of the Cydem VT System to Current Technology across Various CHO Media
- Vi-CELL BLU Analyzer Data Exports and Offline Analysis Instructions
- Use Machine Learning Algorithms to Explore the Potential of Your High Dimensional Flow Cytometry Data Example of a 20–color Panel on CytoFLEX LX
- How to use R to rewrite FCS files with different number of channels
- A new approach to nanoscale flow cytometry with the CytoFLEX nano analyzer
- CytoFLEX nano Flow Cytometer: the new frontier of nanoscale Flow Cytometry
- Detecting Moisture in Hydraulic Fluid, Oil and Fuels
- Detection of Coarse Particles in Silica Causing Cracks in Semiconductor Encapsulants
- Detection of foreign matter in plating solution using Multisizer4e
- Determination of drug-resistant bacteria using Coulter counters
- Determination of Size and Concentration of Particles in Oils
- DO-controlled fed-batch cultivation in the RoboLector®
- dsDNA Quantification with the Echo 525 Liquid Handler for Miniaturized Reaction Volumes, Reduced Sample Input, and Cost Savings
- Screening of yeast-based nutrients for E. coli-based recombinant protein production using the RoboLector Platform
- E. coli fed-batch cultivation using the BioLector® Pro
- Effective Miniaturization of Illumina Nextera XT Library Prep for Multiplexed Whole Genome Sequencing and Microbiome Applications
- Efficient clone screening with increased process control and integrated cell health and titer measurements with the Cydem VT Automated Clone Screening System
- Efficient Factorial Optimization of Transfection Conditions
- Enhancing Vaccine Development and Production
- Enumeration And Size Distribution Of Yeast Cells In The Brewing Industry
- Evaluation of Instrument to Instrument Performance of the Vi-CELL BLU Cell Viability Analyzer
- Filling MicroClime Environmental Lids
- Flexible ELISA automation with the Biomek i5 Workstation
- Friction Reduction System High Performance
- Fully Automated Peptide Desalting for Liquid Chromatography–Tandem Mass Spectrometry Analysis Using Beckman Coulter Biomek i7 Hybrid Workstation
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Get Control in GMP Environments
- Getting Started with Kaluza: Data Scaling and Compensation Adjustment
- Getting Started with Kaluza: Parameters
- g-Max: Added Capabilities to Beckman Coulter's versatile Ultracentrifuge Line
- A method of grading nanoparticles using ultracentrifugation in order to determine the accurate particle diameter
- Compensation Setup For High Content DURAClone Reagents
- HIAC Industrial – Our overview solution for fluid power testing for all applications
- High throughput cultivation of the cellulolytic fungus Trichoderma reesei in the BioLector®
- High-Throughput qPCR and RT-qPCR Workflows Enabled by Echo Acoustic Liquid Handling and NEB Luna Reagents
- A Highly Consistent BCA Assay on Biomek i-Series
- A Highly Consistent Bradford Assay on Biomek i-Series
- A Highly Consistent Lowry Method on Biomek i-Series
- Highly Reproducible Automated Proteomics Sample Preparation on Biomek i-Series
- High-throughput IgG quantitation platform for clone screening during drug discovery and development
- High-throughput Miniaturization of Cytochrome P450 Time-dependent Inhibition Screening Using the Echo 525 Liquid Handler
- Cell Line Development – Hit Picking
- Host Cell Residual DNA Testing in Reduced Volume qPCR Reactions Using Acoustic Liquid Handling
- Exosome-Depleted FBS Using Beckman Coulter Centrifugation: The cost-effective, Consistent choice
- How Violet Side Scatter Enables Nanoparticle Detection
- Automating the Cell Line Development Workflow
- ICH Q2 – the Challenge of Measuring Total Organic Carbon in Modern Pharmaceutical Water Systems
- ICH Q2 – The Challenge of Measuring Total Organic Carbon in Modern Pharmaceutical Water Systems
- ICH Q2 – the Challenge of Measuring Total Organic Carbon in Modern Pharmaceutical Water Systems
- Illumina Nextera Flex for Enrichment on the Biomek i7 Hybrid Genomics Workstation
- Improved data quality of plate-based IgG quantification using Spark®’s enhanced optics
- Increased throughput for IgG quantification using Valita Titer 384-well plates
- Integration of the Vi-CELL BLU Cell Viability Analyzer into the Sartorius Ambr® 250 High Throughput for automated determination of cell concentration and viability
- Temperature dependence of hydrodynamic radius of an intrinsically disordered protein measured in the Optima AUC analytical ultracentrifuge.
- Introducing the Cydem VT System: A high-throughput platform for fast and reliable clone screening in CLD
- Issues with Testing Jet Fuels for Contamination
- Jurkat Cell Analyses Using the Vi-CELL BLU Cell Viability Analyzer
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Linearity of BSA Using Absorbance & Interference Optics
- Long Life Lasers
- LS 13 320 XR: Sample Preparation - How to measure success
- Beckman’s LS 13 320 XR Vs. Malvern Mastersizer
- Machine Learning Algorithms으로 Cellular Subset Composition 의 깊은 insights를 제공합니다.
- Matching Cell Counts between Vi–CELL XR and Vi–CELL BLU
- Media optimization in the RoboLector platform for enhanced protein production using C. glutamicum
- MET ONE 3400+ LDAP & Active Directory connection Guide
- Method for Determining Cell Type Parameter Adjustment to Match Legacy Vi CELL XR
- Migration of Panels Designed on the CytoFLEX S Flow Cytometer to CytoFLEX SRT Cell Sorter
- Miniaturization of an Epigenetic AlphaLISA Assay with the Echo Liquid Handler and the BMG LABTECH PHERAstar FS
- Miniaturization and Rapid Processing of TXTL Reactions Using Acoustic Liquid Handling
- Miniaturized Enzo Life Sciences HDAC1 Fluor de Lys Assays Using an Echo Liquid Handler Integrated in an Access Laboratory Workstation
- Miniaturized Enzymatic Assays with Glycerol
- Miniaturized EPIgeneous HTRF Assays Using the Echo Liquid Handler
- Miniaturized Gene Expression in as Little as 250 nL
- Miniaturized Genotyping Reactions Using the Echo Liquid Handler
- Miniaturized Multi-Piece DNA Assembly Using the Echo 525 Liquid Handler
- Miniaturized Sequencing Workflows for Microbiome and Metagenomic Studies
- Minimizing process variability in the manufacturing of bottled drinking water
- Mixed Mode Sorting on the CytoFLEX SRT
- Mode of operation of optical sensors for dissolved oxygen and pH value
- Modular DNA Assembly of PIK3CA Using Acoustic Liquid Transfer in Nanoliter Volumes
- Multi-Wavelength Analytical Ultracentrifugation of Human Serum Albumin complexed with Porphyrin
- Nanoliter Scale DNA Assembly Utilizing the NEBuilder HiFi Cloning Kit with the Echo 525 Liquid Handler
- Nanoscale Sorting with the CytoFLEX SRT Cell Sorter
- What to do now that ACFTD is discontinued
- Low-pH profiling in µL-scale to optimize protein production in H. polymorpha using the BioLector
- Optimized NGS Library Preparation with Acoustic Liquid Handling
- Particle Counting in Mining Applications
- Performance of the Valita Aggregation Pure assay vs HPLC-SEC
- Phototrophic cultivation of Chlorella vulgaris in the BioLector XT microbioreactor
- Plate Deposition Speed Comparison of Astrios and CytoFLEX SRT Cell Sorters
- Precision measurement of adipocyte size with Multisizer4e
- Principles of Continuous Flow Centrifugation
- Flow Cytometric Approach to Probiotic Cell Counting and Analysis
- Protocols for use of SuperNova v428 conjugated antibodies in a variety of flow cytometry applications
- Purifying High Quality Exosomes using Ultracentrifugation
- Purifying viral vector with VTi 90 rotor and CsCl DGUC
- JP SDBS Validation
- USP System Suitability
- Calibrating the QbD1200+ TOC Analyzer
- Quality Control of Anti-Blocking Powder Particle Size
- Using the Coulter Principle to Quantify Particles in an Electrolytic Solution for Copper Acid Plating
- A Rapid Flow Cytometry Data Analysis Workflow Using Machine Learning- Assisted Analysis to Facilitate Identifying Treatment- Induced Changes
- Rapid Measurement of IgG Using Fluorescence Polarization
- Rapid Rabbit IgG Quantification using the Valita Titer Assay
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Root Cause Investigations for Pharmaceutical Water Systems
- Screening yeast extract to improve biomass production in acetic acid bacteria starter culture
- Single Cell Sorting with CytoFLEX SRT Cell Sorter
- Unveiling the Hidden Signals: Overcoming Autofluorescence in Spectral Flow Cytometry Analysis
- Leveraging the Vi-CELL MetaFLEX for Monitoring Cell Metabolic Activity
- Specification Comparison of Vi–CELL XR and Vi–CELL BLU
- Spectral Flow Cytometry: A Detailed Scientific Overview
- Specifying Non-Viable Particle Monitoring for Aseptic Processing
- A Standardized, Automated Approach For Exosome Isolation And Characterization Using Beckman Coulter Instrumentation
- Streamlined Synthetic Biology with Acoustic Liquid Handling
- Switching from Oil Testing to Water and back using the HIAC 8011+ and HIAC PODS+
- SWOFF The unrecognized yet indispensable sibling of FMO
- Advanced analysis of human T cell subsets on the CytoFLEX flow cytometer using a 13 color tube-based DURAClone dry reagent
- The scattered light signal: Calibration of biomass
- Comparative Performance Analysis of CHO and HEK Cells Using Vi-CELL BLU Analyzer and Roche Cedex® HiRes Analyzer
- Using k-Factor to Compare Rotor Efficiency
- Utilization of the MicroClime Environmental Lid to Reduce Edge Effects in a Cell-based Proliferation Assay
- Validation of On-line Total Organic Carbon Analysers for Release Testing Using ICH Q2
- Vaporized Hydrogen Peroxide Decontamination of Vi–CELL BLU Instrument
- Vertical Rotor Case Study with Adenovirus
- Vesicle Flow Cytometry with the CytoFLEX
- Automating the Valita Titer IgG Quantification Assay on a Biomek i-Series Liquid Handling System
- Evaluating Clone Performance and Cell-Specific Productivity: Comparing the Cydem VT System and 10 L Bioreactor Cultivations
- Rapid, Automated Purification of Adeno-Associated Virus using the OptiMATE Gradient Maker
- Reducing Variability and Hands-On time in Viral Vector purification using the OptiMATE Gradient Maker
- Variability Analysis of the Vi-CELL BLU Cell Viability Analyzer against 3 Automated Cell Counting Devices and the Manual Method
- Automating the Valita Aggregation Pure Assay on a Biomek i-Series Liquid Handler
- Vi-CELL BLU Regulatory Compliance - 21 CFR Part 11
- Whole Genome Sequencing of Microbial Communities for Scaling Microbiome and Metagenomic Studies Using the Echo 525 Liquid Handler and CosmosID
- Sorting Rare E-SLAM Hematopoietic Stem Cells Using CytoFLEX SRT and Subsequent Culture
- Unlocking Insights: The Vital Role of Unmixing Algorithms in Spectral Flow Cytometry
- Vi-CELL BLU FAST Mode Option
- Viral Vector Purification with Ultracentrifugation
- Analytical Ultracentrifugation (AUC) for Characterization of Lipid Nanoparticles (LNPs): A Comprehensive Review
- Leveraging Analytical Ultracentrifugation for Comprehensive Characterization of Lipid Nanoparticles in Drug Delivery Systems
- Catalogs
- Experimental Protocols
-
Brochures, Flyers and Data Sheets
- Access Single Robot System for Synthetic Biology Workflows
- Automated Solutions for Cell Line Development
- Automated Solutions for ELISA
- Echo Acoustic Liquid Handling for Synthetic Biology
- HIAC 8011+ Liquid Particle Counting Systems
- LS 13 320 XR - Laser Diffraction Particle Size Analyzer
- Download the Valita Titer Assay Brochure
-
Case Studies
- Achieving Increased Efficiency and Accuracy in Clinical Testing
- Algae Biofuel Production
- Adenoviral Vectors Preparation
- Antibody and Media Development
- Choosing a Tabletop Centrifuge
- DNA Extraction from FFPE Tissue
- English Safety Seminar
- Equipment Management
- Exosome Purification Separation
- Fast, Cost-Effective and High-Throughput Solutions for DNA Assembly
- High-throughput next-generation DNA sequencing of SARS-CoV-2 enabled by the Echo 525 Liquid Handler
- Membrane Protein Purification X Ray Crystallography
- Organelles Simple Fractionation
- Sedimentary Geology
- Tierra Biosciences reveals major molecular discovery
- University Equipment Management
- Autophagy
- B Cell Research
- Basic Research on Reproductive Biology
- Cardiovascular Disease Research
- Cell Marker Analysis
- Collagen Disease Treatment
- Contribute To Society By FCM
- Controlling Immune Response
- Creating Therapeutic Agents
- Current Status of Cocktail Antibody Reagent Utilization
- DxFLEX that provides On-site Service Support
- Future of Fishing Immune Research
- Hematopoietic Tumor Cells
- Hiroshima Genbaku HP Hematopoietic Tumor Testing
- Improving Efficiency in Clinical FCM Workflow
- Looking to the Future of Research Support
- Opening New Possibilities for Extracellular Protein Degradation
- The Importance of FCM education and CytoFLEX
- Nanoflowcytometry for EV research
- iPS Cell Research
- Leveraging acoustic and tip-based liquid handling to increase throughput of SARS-CoV-2 genome sequencing
- Measuring the number of CD34 using AQUIOS
- Particle Interaction
- Quality evaluation of gene therapy vector
- Retinal Cell Regeneration
- Severe Liver Disease Treatment
- Treating Cirrhosis
- Fundamentals of Ultracentrifugal Virus Purification
- Flyers
-
Interviews
- Background and Current Status of the Introduction of Flow Cytometers
- Bacteriological-measurements-of-soil-bacteria-in-paddy-fields
- Benefits-of-the-coulter-principle-in-the-manufacturing-for-ips-cell-derived-natural-killer-cells
- Breakthrough Solutions for Accurate Microbial Volume and Cell Count Measurement
- Fundamentals of Ultracentrifugal Virus Purification
- Central Diagnosis in the Treatment of Childhood Leukemia 1
- Central Diagnosis in the Treatment of Childhood Leukemia 2
- How the BioLector XT Microbioreactor and Biomek Liquid Handler solutions are transforming precision fermentation
- Challenges-in-viability-cell-counting
- Contribution of Cytobank to 1-cell analysis of the cancer microenvironment
- Development of technology for social implementation of synthetic biology
- Flow Cytometry Testing in Hospital Laboratories
- Fundamentals of Ultracentrifugal Virus Purification
- Tumor Suppressor Gene p53 research and DNA Cleanup Process
- Dr Yabui UCF Lecture
- Importance of Cell Cluster Volume Measurement in Regenerative Medicine
-
Posters
- Applications of Ultracentrifugation in Purification and Characterization of Biomolecules
- Automated DNA Extraction with GenFind V3 on Biomek i7 Hybrid
- ABRF 2019: Automated Genomic DNA Extraction from Large Volume Whole Blood
- Automated library preparation for the MCI Advantage Cancer Panel at Miami Cancer Institute utilizing the Beckman Coulter Biomek i5 Span-8 NGS Workstation
- Automating Cell Line Development for Biologics
- Cell-Line Engeneering
- Characterizing the Light-Scatter Sensitivity of the CytoFLEX Flow Cytometer
- AACR 2019: Isolation and Separation of DNA and RNA from a Single Tissue or Cell Culture Sample
- Preparing a CytoFLEX for Nanoscale Flow Cytometry
- A Prototype CytoFLEX for High-Sensitivity, Multiparametric Nanoparticle Analysis
- ABRF 2019: Simultaneous DNA and RNA Extraction from Formalin-Fixed Paraffin Embedded (FFPE) Tissue
- Quantification of AAV Capsid Loading Fractions: A Comparative Study
- Using Standardized Dry Antibody Panels for Flow Cytometry in Response to SARS-CoV2 Infection
- Product Instructions
-
Whitepapers
- Algorithmic Tools for CytoFLEX mosaic Spectral Flow Cytometer
- Evaluating mRNA-LNP using AUC
- Centrifugation is a complete workflow solution for protein purification and protein aggregation quantification
- Analytical Ultracentrifugation: A Versatile and Valuable Technique for Macromolecular Characterization
- Addressing issues in purification and QC of Viral Vectors
- AUC Insights - Analysis of Protein-Protein-Interactions by Analytical Ultracentrifugation
- Elevate Your Extracellular Vesicle (EV) Research – An Introduction to EVs
- Enhancing Molecular Studies with Multiwavelength Analytical Ultracentrifugation
- GMP Cleanrooms Classification and Routine Environmental Monitoring
- AUC Insights - Assessing the quality of adeno-associated virus gene therapy vectors by sedimentation velocity analysis
- A General Guide to Lipid Nanoparticles
- Analyzing Biological Systems with Flow Cytometry
- Changes to USP <1788> Subvisible Particulate Matter
- Automation Approach to Accelerate Antibody Drug Development
- Characterization of RNAdvance Viral XP RNA Extraction Kit using AccuPlex™ SARS–CoV–2 Reference Material Kit
- CytoFLEX Platform Gain Independent Compensation Enables New Workflows
- CytoFLEX Platform Flow Cytometers with IR Laser Configurations: Considerations for Red Emitting Dyes
- Evaluation of the Analytical Performance of the AQUIOS CL Flow Cytometer in a Multi-Center Study
- Simultaneous Isolation and Parallel Analysis of gDNA and total RNA for Gene Therapy
- Hydraulic Particle Counter Sample Preparation
- Purification of Biomolecules by DGUC
- Beckman Coulter Viral RNA 추출 용해 완충액을 사용한 COVID–19 질병 바이러스 SARS–CoV-2 불활화
- Tips for Cell Sorting
- AUC Insights - Sample concentration in the Analytical Ultracentrifuge AUC and the relevance of AUC data for the mass of complexes, aggregation content and association constants
- Liquid Biopsy Cancer Biomarkers – Current Status, Future Directions
- MET ONE 3400+ IT Implementation Guide
- Reproducibility in Flow Cytometry
- SuperNova v428: New Bright Polymer Dye for Flow Cytometry
- SuperNova v428: New Bright Polymer Dye for Flow Cytometry
- Japan Document
-
어플리케이션 노트
